Final report for GNE18-176
Project Information
Plant viruses cause considerable economic and yield losses worldwide, and since it is not possible as of now to effectively “cure” infected plants, efforts are made into controlling their transmission and spread. However, virus detection, the first step in disease management, sometimes requires extensive training as well as use of bulky or expensive equipment, which can become a burden for farmers and/or extension agents and demand a considerable amount of time. Fortunately, isothermal molecular techniques that can circumvent these issues while maintaining the high standards of sensitivity and specificity, are becoming more and more popular. Recombinase Polymerase Amplification is a molecular technique that is comparable to the gold standard of molecular detection that is the Polymerase Chain Reaction (PCR). RPA provides the same sensitivity and specificity as PCR while also allowing for speediness of results interpretation and more streamlined. We describe hereby how this technique was developed and used for detection of the plant virus Tomato spotted wilt orthotospovirus, as well as three different but complementary ways of interpreting results both in end-point (after the reaction is complete) and real-time (while the reaction is happening) and compatible with several equipment.
Hypothesis: A recombinase Polymerase Amplification (RPA) can be engineered for on-field detection of Tomato spotted wilt virus with great sensitivity and specificity. Additionally the use of the 3D nanotube-based filter CNT-STEM will allow an increased detection and reliability for the detection technique. By providing our field-friendly virus-capture device and combining it with RPA that doesn’t need bulky and non-portable laboratory equipment, we expect to provide farmers and field pathologists with a system that allows early on-field virus detection, has a great sensitivity and does not require professional training or shipment to a laboratory.
Specific objectives are:
1. To develop and validate a RPA reaction for detection of Tomato spotted wilt virus
2. To integrate CNT-STEM as a virus capture platform with the molecular detection of genomic fragments of RPA in asymptomatic infected tomato seedlings or just-virus-inoculated tomato plants
3. To compare the detection of TSWV in RPA-only and RPA+CNT-STEM for field samples
Plant viruses are a threat to food security, because of the huge amount of losses (sometimes as high as 100% for Tomato spotted wilt otrhotospovirus (TSWV)) they cause on staple crops; as intracellular parasites, no prophylactic measures are available for treating plants other that controlling their vector for transmission to new hosts (Nicaise 2014)
SARE reports for projects LS07-199 (Southern SARE), SW12-040 (Western SARE) and OW14-026 (Western SARE) show that there has been interest in finding solutions for plant viruses related to the tomato production and all fall into the specifications and requirements of sustainable agriculture, as Research and education and professional producer components.
Spotted wilt of tomatoes is a devastating plant disease that causes big losses for the tomato industry (Sevik and Arli-Sokmen 2012). This disease is caused by TSWV, a plant virus with a very wide host range and transmitted (or vectored) by at least nine different species of tiny thrips insects. Even though countermeasures are exercised to ensure the confinement of the virus and the control of its vectors, the tomato industry and smaller growers are still trying hard to minimize the losses to the virus. Therefore, this important virus can be used as test-case for other viruses infecting tomatoes.
Surveys conducted with four Pennsylvania farmers in April of 2018 showed that viral diseases, especially TSWV are a concern in the state (growers responded to be from moderately to very concerned), farmers apply management strategies for managing thrips (3 out of 4 do not apply pesticides), and 3 out of 4 work with specialists and extension agents to make important decisions for their crops. All of the interviewed farmers agreed that they would be interested to try our proposed technology and interestingly, they would prefer a technology for detecting the viruses themselves in the field.
Laboratory techniques can be expensive and labor intensive, and in the end can fall short on detecting low titer viruses. The Recombinase Polymerase Amplification or RPA detection system proposed by us is portable, affordable and can give a result in as short as 30 minutes. If further validation is needed, the products of RPA can undergo the same downstream applications as any amplification reaction. RPA produces a huge number of copies of the virus genomic region and renders the presence of the viral genome visible via a portable amplification detection chamber.
The direct link of this project towards sustainable agriculture is to enhance the quality of life for farmers and society and ensure food security. By allowing an on-site detection, farmers and extension agents can test for virus-infected plants before they become foci of infection, additionally weeds can be tested for making sure they are not reservoirs of disease and better-integrated-pest-management (IPM) strategies can be performed.
Moreover, our lab has been pioneering the use of a microfluidics device know as CNT-STEP (Carbon nanotube tunable enrichment platform) for the physical entrapment of viruses and the increase in limits of detection as described in previous systems and publications (ref). The increase in accuracy and timeliness of our proposed device and procedure should allow a grower or extension agent to decide confidently that a pesticide to control vectors is or not needed. Reducing pesticides will have significant economic, environmental and human health benefits. Earlier pathogen detection will allow earlier management decisions, which should reduce pesticide applications. Additionally, since the diagnosis is made in the field, the costs of sending samples to a lab could be reduced and limited to those samples that need further review or specific characterization.
Cooperators
- (Researcher)
- (Researcher)
- (Educator and Researcher)
Research
Nicothiana benthamiana plants for the experiment were reared for a total of 3 weeks (1 week as a seedling, 2 weeks after transfer to pots). Plants were inoculated with roughly 1g of TSWV-Chrys5-infected plant tissue with inoculation buffer, alongside Carborundum and activated charcoal. Emilia sonchifolia plants were also used and reared as described for the N. benthamiana All homogenization procedures were made using roughly a leaf, homogenized into phosphate buffered saline (PBS) using a disposable extraction bag and a hand homogenizer; 7 dpi and kept at room temperature. This extract was serially diluted up to 10^-20, by mixing 20 μl of solution and 180 μl of nuclease-free water and repeating this 10 times.
We modified the protocols of commercially available kits that are used for DNA amplification via RPA to amplify RNA instead, since TSWV, as most plant viruses, possesses a genome composed of RNA. For this purpose, we used the AmplifyRP Acceler8 kit (Agdia, Inc., Elkhart, IN) and added the iScript™ Reverse Transcription Supermix (Bio-Rad, Hercules, CA) for converting the RNA of TSWV into complementary DNA (cDNA). We also designed original and novel primers and a probe (both being small sequences of DNA, artificially synthesized that bind to the virus target genomic region to ensure unambiguous detection of the target virus) specific for amplification of a portion of the TSWV genome.
The goal of the RPA is to make a huge number of copies of the virus genomic region contained between the primers in a small 1.5 ml test tube. This amplification renders the presence of the viral genome visible via a portable amplification detection chamber, sold separately (Agdia, Inc., Elkhart, IN). The portable chambers are plastic snap-on chambers that smash the reaction tube alongside a reaction mix and force it through a lateral immunostrip that detects the presence of the amplicon. The immunostrip gives a positive result if the control band and the test band are visible, negative if only the control band is visible, and invalid if there are no bands. However, this ‘amplification’ and ‘visualization’ will only happen if the sequence designed as the target is present, therefore it can be used as a useful detection tool (Figure 1).
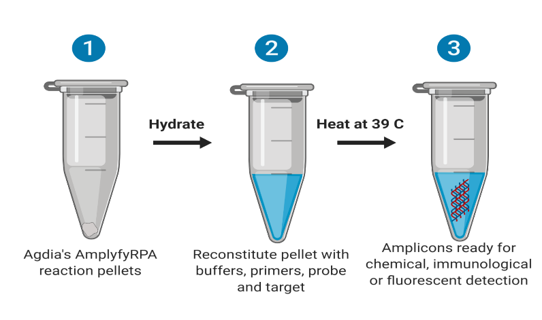 Figure 1: Overview of the reaction. Reproduced from Iturralde-Martinez and Rosa (2019).
Figure 1: Overview of the reaction. Reproduced from Iturralde-Martinez and Rosa (2019).
The sequences for the primers and probe and the modifications made to them are shown below (Iturralde-Martinez, 2018):
End-point probe: 5′-/56-FAM/AT TGT ACA AAG GTT TGT TTC GGA ATA AAT CTA GG /idSp/AT TCG CAA CCT AAT CT/3SpC3/-3′
Real time probe: 5’-ATTGTACAAAGGTTTGTTTCGGAATAAATC/iFluorT/AGG/idSp/AT/iBHQ-1dT/CGCAACCTAATCT/3SpC3/-3’
Reverse: 5′-/5BiosG/ATATTGTTATAGAAGGTCCTAATGATT-3′
Forward: 5'- GAATCTATTATACCATTTCTCAATCTCTTAGC -3'
From which: 56 FAM and IFluorT are fluorescein molecules located at respectively the beginning or in the internal part of the oligo, idSp is a spcer or modified base that helps trigger recombination, 3SpC3 is a spacer at the 3′ end of the oligo and 5BiosG is a residue with a biotin molecule attached and iBHQ is an internal Black hole Quencher that keeps the internal fluorescein quenched until activated. Two species of amplicons are generated, one that is flanked by the primers Forward and Reverse and one between the Probe and the Reverse primer, being doubly marked with FAM at its 5′ end and biotin at its 3′ end.
For performing the reaction, according to the manufacturer’s procedure: 5.75 μl of rehydration buffer; 0.5 μl of each primer and 0.25 μl of the respective probe, all in concentrations of 10 μM; 0.50 μl of 280 mM magnesium acetate (MgOAc) and 0.25 μl iScript™ Reverse Transcriptase (Bio-Rad, Hercules, CA) were mixed with a 2 μl of crude plant sap and with a reaction pellet (provided in the kit). Water was added up to a total volume of a reaction of 10 μl, and the mix was transferred to a 0.2 mL microfuge tube. Upon closing the vessel, the reaction was maintained at 39 ⁰C on a portable thermal block for 20 min, and the results interpreted afterward. For non-target controls, water was used instead of the plant extract.
For cheaper in field visualization methods, we also modified and optimized the method originally published by Koo, Wee et al. (2016) that consists of adding 1.8 μl of magnetic beads to the 10 μl reaction according to their manufacturer’s protocol. These beads bind exclusively to newly synthesized DNA and thus can be added to any product of DNA amplification. After recalling the beads with a strong magnetic rack, the supernatant is discarded and replaced by a flocculation agent like magnesium acetate (MgOAc); in those reactions that are positive (amplicon present), floccules of beads, DNA and the flocculent agent sinks to the bottom of the vessel, making the entire mix clear. In those samples that do not have an amplicon the recalled beads re-dissolve in the liquid, causing it to be brown.
As an extra assay, we used the same primers with the real-time probe and tested it for real time detection of amplification. Since we did not possess a portable fluorometer as suggested by the manufacturer, we used instead our Real Time PCR thermal Cycler. See Figure 2 for a summary of the mode of action of the described methods (Figure 2).
.
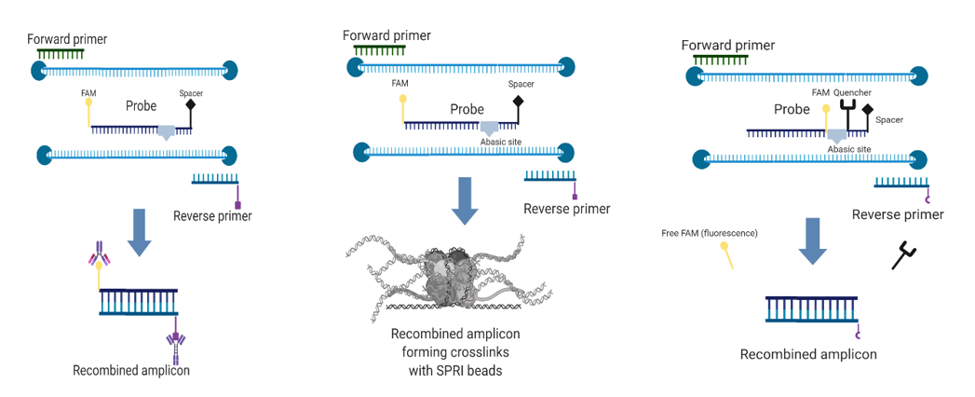
Figure 2: Modes of action of the two end-point assays (left and center) and the real-time assay. Reproduced from Iturralde-Martinez and Rosa (2019).
Quantification of starting viral material
An RNA extraction optimized for liquid samples was performed on 50 μl of undiluted extract and used for real-time PCR amplification as described before. The reaction was made in triplicate and the obtained average Cq was interpolated into the standard curve for approximating the number of viral copies (TSWV’s genomic segments).
Sensitivity
For testing sensitivity, as explained before, the extract was serially diluted into a final volume of 200 μl, having an aliquot volume of 20 μl, this was done successively until reaching 10^-20 dilution. Then each 10^-4 multiple dilution was tested in parallel using end-point RPA+ Amplification Detection Chambers, end-point RPA+ flocculation assay and finally RPA in real time in the CFX96 thermal cycler.
As shown in the picture (Figure 3), only the reaction mix that contained the sap from the TSWV infected plant (right chamber) developed two bands in the amplification detection chamber, indicating a positive test. The test line is made by antibodies that detect the doubly marked (5′ FAM and 3′ biotin) amplicon and the control line contains antibodies that react to the reaction mix.

Figure 3. Detection of the amplicon using official Agdia Amplification detection chambers. Left: Negative result, only the control line is visible. Right: Positive result: Both the control and test lines are visible.
The image (Figure 4) shows results from the same RPA reaction mix now combined with the SPRI beads:

Figure 4. Detection of the amplicon using SPRI beads. Left: Positive result. the amplicon makes a stable precipitate with the beads, resulting in a clear solution. Center and right: Negative result in which the beads are spread throughout the mix.
In the case of the beads, the results were consistent with those from the Amplification detection chambers, in fact, the same plant sap that produced a double band (positive result) had a precipitate (positive result). The precipitate was stable and did not dissolve back into the reaction, confirming the presence of the amplicon by two methods.
The RPA reaction was successful in every case. For the end-point cases, the results were interpreted once the reaction was finished, 30 minutes after mixing the components and keeping it at 39 ̊C; per manufacturer recommendations, the first method for confirming the presence of the produced amplicon was to use the proprietary Amplification Detection Chambers. The way these chambers operate is property of AGDIA; nonetheless, the basic interpretation is to visualize one line on the control (C) field to determine a test as valid and another line on the test field (T) to test for the presence of the tagged amplicon. For our case, the case of an infected plant was visualized as a chamber with two lines, whereas for the negative controls, an uninfected plant and a reaction without template showed both only the control line.
Afterward, the same reaction was verified by the flocculation method in end-point as well, meaning after the reaction has been completed. For the cases presented, it was possible to see that the reaction that contained extract from an infected plant presented a stable precipitate at the bottom of the vessel; meanwhile the controls remained opaque and did not show any detectable pellet.
Finally, for the extra assay of using a real time probe and following the reaction as it progresses using a optically enabled Real Time PCR thermal cycler, we were able to obtain an amplification curve for only the experiment that had extract from an infected plant, the other two controls appear in the amplification plot as horizontal lines that overlap with the x axis (Figure 5).
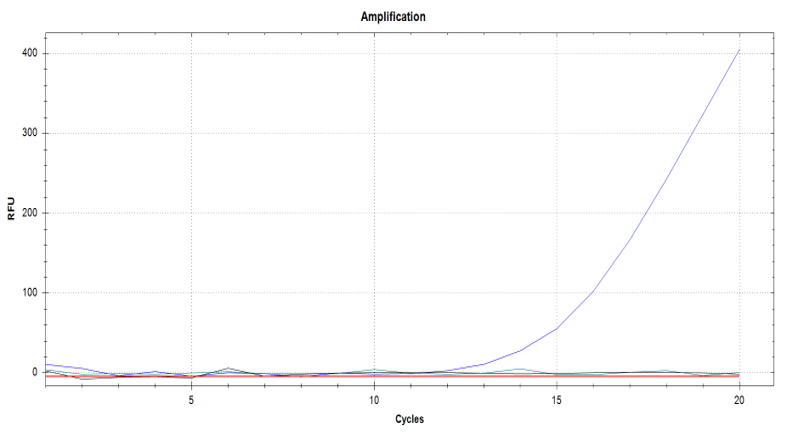
Figure 5. Amplification curves for the real-time RPA experiment. The only visible curve is the positive result, corresponding to the reaction that contained crude plant extract from an infected plant. Negative controls are parallel to x-axis. Extracted from Martinez & Rosa, (2019).
Sensitivity assay
The serial dilutions were assayed in multiples of 10^-4 using the two described methodologies for detection of amplicon in end point and real-time. For the first case, we used dilutions ranging from 10^-4 to 10^-20; and including as a positive control a crude, undiluted plant extract and as negative controls a healthy plant extract as well as a non-target control using water instead of plant extract. Using the Amplification Detection Chambers, the highest dilution in which amplification was detected was the 10^-12 dilution, approximated by standard curve to have about 1 genomic copy of the virus present.
The same serial dilutions range was tested in end-point with the flocculation assay in parallel. For this case, the results were as expected, there was a visible focus from the dilution 10^-4 to the dilution 10^-12 (~1 copy); further dilutions did not present a visible pellet nor a clarified buffer and were not visually different from negative controls. Because our RPA results gave a positive result with as low as one copy of TSWV (one virus), we could not use the CNT-STEP devices to improve detection below the limit of one molecule.
Finally, another replicate of the dilutions set was tested in real time with the modified protocol and visualized with the Real Time PCR thermal cycler. The undiluted, positive control had the highest concentration showing a recognizable amplification curve. The next two dilutions 10^-4 and 10^-8 presented amplification curves as well. Nonetheless, all the dilutions past 10^-12 did not present amplification curves in the allotted time of 20 minutes, unlike the end-point assays previously described in which it was still possible to detect a single copy of the virus. The negative controls did not present amplification curves either.
TSWV isolates specificity assay
In order to determine if the designed RPA would work for more than one isolate of TSWV, the reaction was made with all the isolates our lab possessed, being TSWV-Chrys5, TSWV-Tom2 and TSWV-PA01 isolated from chrysanthemum, tomato and pepper respectively and maintained in N. benthamiana (Chrys5 and Tom2) and Emilia sonchifolia (PA01). The isolates were only tested in endpoint using the two described methodologies above, and we were able to detect all TSWV tested isolates in both plant species.
We were able to design a molecular technique for the rapid and sensitive detection of Tomato spotted wilt orthotospovirus that is portable and can be interpreted using three different tools: amplification detection chambers, SPRI beads + crowding agent and fluorometry, using a modified probe. All these techniques can be used interchangeably for a qualitative detection of TSWV and can be scaled up for lab-on-a-chip applications or point-of-care detection of virus, to make quicker informed decisions in resource-limited environments, and without the need of trained personnel.
RPA is a portable and reliable technique comparable to PCR, that has great sensitivity and specificity and can be used for detection of pathogens both in end-point and real-time. The technique circumvents RNA extraction and reduces the technicality to such a point any trained applicant would be able to perform and interpret it.
The technique was performed in three different ways that all corroborate each other, two in end-point after the amplicon has been made and is either detected with immunology-based techniques (Amplification detection chambers) or via chemical flocculation using magnetic beads. This same amplicon is detected as it is being produced by our real-time version of the technique. This ensures to show a plethora of ways for testing TSWV that can fit different budgets and levels of training, with the same stringency in sensitivity and specificity.
For our present case, the RPA was highly sensitive, being able to detect an approximate of 1 TSWV genomic segment (interpolated using PCR). Moreover, it was possible to detect with the same technique up to three isolates that our lab possessed at the time of testing.
Because of this unique features of RPA, we determined the RPA by itself was optimal for in-situ detection of the virus, with a streamlined procedure that aims to give a quick an reliable detection.
Our results were reported in poster format at the Plant Health Conference. Cleveland, OH. August 2019 (Iturralde-Martinez, Juan Francisco. Rosa, Cristina. In-situ detection of Tomato spotted wilt orthotospovirus from crude plant extracts using Reverse Transcriptase- Recombinase Polymerase Amplification (RT-RPA) in endpoint and real-time) and published in Martinez, J. F. I., & Rosa, C. (2019).
A fact sheet on TSWV and other important tospoviruses was developed and provided to our vegetable pathology extension faculty at Penn State, for publication on the University Extension Website.
References
Iturralde-Martinez, Juan Francisco. Rosa, Cristina. In-situ detection of Tomato spotted wilt orthotospovirus from crude plant extracts using Reverse Transcriptase- Recombinase Polymerase Amplification (RT-RPA) in endpoint and real-time. Poster at Plant Health 2019 Conference. Cleveland, OH. August 2019.
Koo, K. M., et al. (2016). "A simple, rapid, low-cost technique for naked-eye detection of urine-isolated TMPRSS2: ERG gene fusion RNA." Scientific reports 6: 30722.
Martinez, J. F. I., & Rosa, C. (2019). In-situ detection of Tomato spotted wilt orthotospovirus from crude plant extracts using Reverse Transcriptase-Recombinase Polymerase Amplification (RT-RPA) in endpoint and real-time. bioRxiv, 720623.
Nicaise, V. (2014). "Crop immunity against viruses: outcomes and future challenges." Frontiers in plant science 5: 660.
Sevik, M. A. and M. Arli-Sokmen (2012). "Estimation of the effect of Tomato spotted wilt virus (TSWV) infection on some yield components of tomato." Phytoparasitica 40(1): 87-93.
Education & outreach activities and participation summary
Participation summary:
We initially surveyed four vegetable farmers to determine the need for our research.
The methods described in this project as well as a detailed description of results have been compiled into a journal article that is as of now in a online article hereby attached. This preprint have been submitted to the Journal Scientific Reports.
Additionally, we made a fact sheet indicating important facts and recommendations about our model virus TSWV and a closely related virus called INSV that is economically important as well and which can be practically indistinguishable from TSWV without specialized equipment.
Project Outcomes
Plant viruses are a threat to food security, because of the huge amount of losses (sometimes as high as 100% for TSWV case) they cause on a series of staple crops. Unfortunately, control of viruses in plants is based on insecticide sprays aimed at controlling the insects that vector those viruses. We are here providing a tool that will allow growers to detect viruses earlier or and more accurately, and will hopefully reduce the amount of insecticides used as preventative measures in the fight against plant viruses. By allowing an on-site detection, farmers and extension agents can test for virus-infected plants before they become foci of infection, additionally weeds can be tested for making sure they are not reservoirs of disease and better-integrated-pest-management (IPM) strategies can be performed.
Reducing pesticides will have significant economic, environmental and human health benefits. Additionally, since the diagnosis is made in the field, the costs of sending samples to a lab could be reduced, and limited to those samples that need further review or specific characterization.
This project in the future will help to increase the quality of life of farmers and final consumers of tomato, as well as a way to ensure food security and save farmers money by being able to make detection by themselves and in less than one hour.
Even though our outcomes could not be directly extrapolated to the field and to change and improve the lives of farmers, research that aims for easier and meaningful detection methods for plant pests has the potential of shortening the gap between scientists and farmers. By providing rigorous methods that extrapolate lab science to field, we expect to see more of this in the upcoming years.
Through this project we established a routine collaboration with our collaborators that are specialists in integrated pest management and sustainable agriculture. Their input provided more awareness about sustainable agriculture for the students and PI and other collaborator involved in this project. The Pi also was invited to participate in the Online Conference Northeastern IPM center by Cornell University, establish to increase knowledge about IPM and sustainable agriculture for the scientific community.
The publication on the Penn State Extension website of our factsheet will be an important step to ensure that farmers in Pennsylvania are more aware and educated about viruses, especially those that cause the most economical losses.
Hopefully, extension will pick up on isothermal detection methods, as well as the methods will get easier and more streamlined to be used by the growers and extension managers.