Final report for GNE16-117
Project Information
The objective of this project is to provide poultry farmers and industry workers with preliminary insight into the effects of incorporating food scraps into an on-farm composting system. The primary goal is to perform shotgun metagenomic sequencing in order to identify and quantify antibiotic resistance genes (ARGs) present in various products and intermediaries on a poultry farm currently accepting food scraps, ranging from the scraps themselves to final consumer products such as eggs and home gardening supplies. This project emerged in response to new legislation, Act 148 in Vermont, which dictates the diversion of food scraps for agricultural practices such as poultry feed. However, the persistence of resistance genes throughout this cycle is currently unknown and may pose a risk in the burgeoning fight against global antibiotic resistance. Additionally, on integrative farms such as the one sampled here, the potential for introduction of novel resistance (human-associated) into the farm setting and amplification of resistance through the spread back to consumers the need for investigations such as this is paramount.
In order to accomplish the aims of quantifying antimicrobial resistance genes, a relatively small subset of genetic material contained within compost, a total of 14 samples were sequenced in duplicate. This allowed both an increase in read depth and probability of finding targets of interest. Additionally, the comprehensive approach to sampling across the farm identified ecological niches on the farm where both microbiomes and resistomes appeared most similar. Sampling occurred in February 2017, which was delayed due to severe cold that impacted the farm’s ability to collect and properly rinse food waste containers. Samples included four off-site commercial facilities, nine on-farm sites, and a trip/reagent blank to allow for bioinformatic filtering of false positive results. Due to the novelty of this project, we adapted previous methodologies for DNA extraction to limit the contamination of samples by plant and human genetic material, which will streamline the process in future studies.
A total of 50 unique resistance genes were found across all 13 samples. While total number of ARGs varied across samples, from 21 in the egg wash to 3 in the grocery store sample, off-farm samples contained ARGs of higher clinical concern and on-farm samples contained mainly ARGs associated first-line antibiotics less commonly used in clinical medicine or commonly associated with soil-dwelling bacteria. Microbial community was also assessed, with distinct patterns on and off-farm, as well as between different areas on the farm (depicted in Figure 1). Human pathogens of concern, such as ESKAPE pathogens, were found in off-farm samples but did not persist through the composting process. Finally, virulence genes were found in all samples that, again, appeared to be mediated by the composting process. Genes common to off-site samples occurred in raw food scraps and composts on the farm, but very few were also present in final composts or commercial products such as eggs and worm castings.
In conclusion, the practice of utilizing food scraps for use as poultry feed and raw materials for compost and worm castings appears to not increase the prevalence of resistance genes or pathogens of concern. Current management practices appear to be sufficient control measures in preventing the spread of novel ARGs, however more farms should be enrolled in surveillance in the future. Finally, we were successful in creating a methodology and reproducible bioinformatic pipeline for the screening of diverted food scraps using shotgun metagenomic sequencing.
Introduction:
In the U.S. poultry industry, there has been a push from consumers, producers, industry stakeholders and legislative powers to pursue sustainable feeding programs. While this represents an important step in the zero waste and sustainability initiatives, the food safety status of these materials has not been fully examined. It has been shown that both resistant microorganisms and ARGs are present in food and food waste samples [7]. However, current detection for food waste diverted to the poultry industry is limited. For more than a decade ARGs have been identified as a source for the rapid proliferation of resistance in the microbial population, a result that merits further investigation. However, current technologies rely on culture dependent or extremely targeted molecular methods both of which are labor intensive and limited in scope. Culture dependent techniques are biased against hard to culture organisms or those of low abundance [6], while targeted molecular methods, RT-qPCR, require previous knowledge of gene sequences and primers. Such screening efforts are limited to the number of primers selected and cannot provide information on new variants or genes present in a sample. A thorough metagenomic sequencing approach will not only rapidly increase knowledge in this area but will influence predictive models on the spread and perseverance of these genes in the farm setting. In the age of environmental sustainability and state-mandated waste reduction, there is a pressing need to characterize these communities and understand the mechanisms behind gene transfer on farms. This proposed research is significant because introduction of metagenomic-based techniques will greatly increase our abilities to characterize ARG diversity in food waste materials. Potential benefits of this work are numerous as antibiotic resistance is an issue on a global level. Availability of a rapid, inexpensive, and widely applicable detection method will allow our knowledge of this issue to increase exponentially and support change on an equally large scale. As manure samples are likely to contain ARGs, they will act as a control for the assessment of ARGs in compost that may be coming from food wastes. For example, genes present in the compost samples that are not present in manures are likely sourced from food wastes. In addition, screening the output, eggs, will allow us to better quantify how these genes may circulate back into the human food system. These data will inform management practices to ensure sustainable use of food wastes, increase livelihood security, allow long-term productivity, and reduce the spread of resistance. The mixing of many food sources may act as a “melting pot” for antimicrobial resistance leading to increased risk of dissemination and transfer. Multidrug resistant (MDR) bacteria, those that are resistant to more than one class of antibiotic, are becoming more common in agriculture and food products, and frequently the multidrug resistant genes are maintained as a cluster or cassette of genes within a mobile genetic element. Consequently, the use of one class of antibiotic can select for and maintain MDR bacteria carrying multiple ARGs. As such, surveillance and limitation of introduction events are just one of many ways we can limit the growth of antimicrobial resistance in agriculture.
Shotgun metagenomic sequencing has been successfully used in other fields for identification of antimicrobial resistance genes (ARGs) and these protocols will be modified to address this project [15]. From monitoring ARGs on paper currency [8], to soil samples from polluted lakes [2], these next-generation techniques have proven to be an invaluable tool. By screening multiple sample types, we can assess variability and dissemination within the system. Applying this cutting-edge technology to food waste recycling businesses that have integrated poultry production will allow comprehensive surveillance at lower cost and faster rates than ever before. To the best of our knowledge, this would be the first project to use these technologies as a screening tool for diverted food wastes and on-farm composts.
While final results were positive, several delays in the project occurred. Firstly, weather conditions delayed sampling as off-farm collection of food scraps required temperatures above freezing for running water to be available on the truck. Even after collection, method development for both pre-processing and DNA extraction required significant trouble-shooting resulting in sequencing being delayed until May 2017. The following information refers to fulfillment of objectives with the modifications made to the original proposal.
- Develop a novel screening method to determine the prevalence of ARGs in unprocessed food waste received by selected poultry farms in Vermont.
Specific aims of this objective include:
- a) Identify target genes of interest for screening and create a comprehensive reference database for sequencing,
- b) Identify target genes with known primers for validation of metagenomic analysis,
- c) Identify antimicrobial resistance genes (ARGs) in food waste samples taken from poultry farms, and
- d) Categorize the types and abundance of ARGs identified in food waste samples obtained from poultry farm(s).
Objectives a, c, and d have been fully completed. The CosmosID bioinformatic suite was used as a reference database and categorization of the resistome of each sample. All samples were sequenced in May 2017 at the Vermont Cancer Center Advanced Genomics core. A total of 13 experimental samples and one control were used, and resistance genes were found in 10 of them. Objective 1b was modified to encompass a more functional component to this study. From the time of the original proposal, it has been more widely accepted in literature to use shotgun sequencing as a tool without validation via qPCR assay. This allowed us to test the phenotypic resistance of microorganisms grown from our samples. At this time, samples have been tested on plates containing two concentrations of Tetracycline. All samples containing Tetracycline resistance genes were able to grow under these conditions, while samples without relevant ARGs were not. This assay system demonstrates that not only are ARGs present in these samples, but they are functional copies as well.
- Quantify the diversity of food waste management practices being implemented on integrated composting and poultry farms in Vermont. In the face of growing antimicrobial resistance concerns, characterizing farmers’ current awareness and efforts to prevent ARG introduction and spread is critical. For the 3 study farms, we will complete a survey to assess the level of knowledge about proper food waste handling and identify measures already being taken to prevent the spread or growth of antimicrobial resistance.
Specific aims include:
- a) Assess the level of knowledge of poultry farmers about limiting antimicrobial resistance and
- b) Identify measures utilized at the farm level to reduce antimicrobial resistance.
Objective 2 has been partially completed. As a single farm was sampled, a comprehensive questionnaire was not applicable. Instead, conversations with the farmer and reporting material was used to gather information about current on-farm management practices. Again, due to the limited literature on this subject, we can only speculate at this time which of the current practices has led to a reduction in ARGs of concern during composting. However, this objective provides critical preliminary data for future studies as well as baseline management practices for farmers newly incorporating food scraps.
Cooperators
Research
- Sample Collection
Samples were collected both on farm and individual food scrap collection sites in February 2017. For each substrate type, four sterile RNA/DNA free 50 mL conical tubes (Ambion) were filled using grab sampling across various depths and locations of on-farm piles or across bins at collection sites. However, due to the time of year, much of the substrate was frozen and this did impact the ability to sample from the core of outdoor samples. For eggs, three eggs were taken directly from hen houses within the barn. All samples were transported on ice back to the University of Vermont and stored at -80 C until further processing.
- Pre-processing
In order to reduce the amount of Eukaryotic DNA contamination, pre-processing via vacuum filtration was performed prior to DNA extraction. Briefly, into four tubes per sample, 1 g of thawed material was added to 10 mL of sterile UltraPure water (ThermoFisher). This was performed for all samples except the egg shell and egg wash. For the egg wash, a whole egg was placed into a sterile Whirl-Pak with 40 mL of UltraPure distilled water warmed to 42° C and gently shaken for 2 minutes. This wash material was then placed into a sterile 50 mL conical for further processing. Once the egg had been washed, they were cracked on the edge of a sterile beaker and all interior products were discarded. Any remaining albumin was rinsed thoroughly with additional sterile water. The shell was then crushed with a gloved hand and inserted into a sterile 50 mL conical tube with 40 mL of warmed (42° C) sterile water and agitated/crushed for 2 minutes with a sterile glass rod adapted from a previous study [13]. From this point, all 50 mL conical tubes (4 x 12 samples, plus 1 x for egg samples) were shaken in a multitube shaker for 5 minutes at 1500 rpm to disrupt bacterial adhesion to any food scrap products. All samples were then filtered through a 40 micron SteriFlip (Millipore) tube using vacuum filtration. This material (now ~40 mL for all samples in a single tube) was then centrifuged for 15 minutes at 2,000 g. Supernatant was discarded and pellets were resuspended in 800 µL of sterile water prior to DNA extraction. Samples were stored at -20° C if not immediately used for extraction.
- DNA Extraction and Quantitation
Due to the novelty of this experiment, literature on DNA extraction for these materials was limited. As such, multiple techniques were employed to achieve the highest quality and quantity of DNA . Three extraction kits were used: QIAmp DNA Microbiome (Qiagen), EZNA Bacterial DNA (Omega), and DNeasy Powersoil (Qiagen, formerly Mo Bio). While the QIAmp and EZNA kits did successfully extract DNA, it was at a much lower concentration than the Powersoil kit. Therefore, the Powersoil kit was used for all future sequencing. Manufacturer’s protocol was followed with the following changes. Briefly, 400 µL of the pre-processed liquid material from each sample was added to a sterile tube containing beads rather than unprocessed soil. Additionally, a negative control for each batch of extractions was included to ensure that there was no contamination at this stage. After extraction samples were stored at -20° C until quantitation and sequencing.
The concentration of DNA in each sample was quantitated using the Qubit 2.0 dsDNA BR Assay system (ThermoFisher). The manufacturer’s protocol was followed and 1 µL of sample DNA to 199 µL of working solution was used. Concentrations ranged from <0.025 ng/µL in the trip blank to 13.5 ng/µL in the finished compost, with an average of 3.7 ng/µL in non-blank samples.
- Shotgun Sequencing
All 14 samples (13 samples + 1 trip blank) were sequenced via 100 bp single end (SE) Illumina HiSeq shotgun sequencing. A total of 2 ng of DNA from each sample was used for library preparation using the Nextera reagent kit. All libraries were checked for quality using the Bioanalyzer system prior to sequencing. Two lanes in total were used, from different flow cells and on different days, as technical replicates as well as increase the total sequencing depth. Library preparation and sequencing was performed at the UVM Cancer Center Advanced Genomics Lab.
- Bioinformatics & Sequence Analysis
Initial sequence analysis was performed by the UVM Bioinformatics core. This included demultiplexing (assigning reads to their sample using the barcodes from the library preparation stage), quality checking using FastQC [Andrews, 2010] and Trimmomatic [Bolger, 2014], and storage on the Blue Moon remote server (VACC). Once sequences were retrieved from the server, quality was examined using FastQC output files. Average sequence length was 120 bp and average quality was above Q 30, indicating that both lanes had high-quality sequences and required no additional processing. Again, due to the novelty of this experiment multiple algorithms for antibiotic resistance gene detection were employed. Prior studies used ARDB and Resqu databases for ARG annotation for analysis [8, 2]. However, source material from these studies were lake sediment and swaps of paper money respectively which likely contain much less Eukaryotic DNA contamination compared to our samples. In order to accurately and efficiently identify both ARGs and bacteria present, two cloud-based systems were used for this project that rely on data mining and k-mer approaches rather than sequence assembly and alignment. This approach allows for better coverage of individual genes given the relatively short sequences generated by shotgun sequencing.
Both One Codex (Reference Genomics, Inc.) [11] and CosmosID (CosmosID Rockville, MD) applications were used for initial sequence analysis. After comparison of output from both systems, final results from only CosmosID will be presented. Briefly, the CosmosID bioinformatics package is a cloud-based platform that uses curated reference datasets to rapidly assign metagenomic reads to the species, sub-species, and even strain level, as well as a wide array of virulence factors, antimicrobial resistance genes, and more. This is accomplished using two main algorithms, the first of which is the ‘pre-computation phase’ which constructs a whole genome phylogeny tree using sets of fixed length n-mers (referred to as biomarkers) from the curated database. Once constructed, the second ‘per-sample phase’ searches metagenomic reads from submitted samples against the biomarker ‘fingerprints’ for identification. Resulting statistics are aggregated to maintain overall precision and allow for sample composition, relative abundance estimates, frequency of a biomarker hit, total coverage of the reference sequence (Total Match %), and total coverage of unique biomarkers (Unique Match %). Total match % is an estimate of total gene or genome coverage, which is used in combination with genome size to calculate relative abundance in order to reduce bias due to the vast differences in microbial genome size. Results from Bacteria Q3 2017, Antibiotic Resistance Q4 2016, and Virulence Factors Q4 2016 were exported in .csv format for additional analysis in RStudio [version 1.0.143]. Filtering of reads was conducted by using all results from the trip/extraction blank (TRBL); previous studies utilizing shotgun metagenomics have noted that reads associated with reagent contamination can occur [ 10, 17]. Briefly, all results that had a match in the TRBL files were removed from further processing. As an additional quality control step, all results that were identified to the same level of “branch” (no specific species or sub-species result) in either TRBL sample were also removed. Effects of this filtering are shown in Table 1.
- Functional Resistance Assays
After successful completion of bioinformatic analysis, an additional functional component was added to this project. The goal of this was to provide evidence that not only were genetic markers of antimicrobial resistance present, but isolates from these samples were functionally resistant as well. To accomplish this, bacteria were isolating and grown on agar media containing one of the following antibiotics: Tetracycline, Ciprofloxacin, Erythromycin, Gentamicin, and Penicillin. These drugs were chosen as they belong the classes of drugs with ARGs found most frequently from the metagenomic data, as well as being widely used in both clinical and agricultural settings.
Bacteria were isolated from the same pre-processed samples used for DNA extraction. Briefly, 50 µL of each sample was added to a Mueller-Hinton agar plate containing: 1) no antibiotic (+ control), 2) Tetracycline, 3) Ciprofloxacin, 4) Erythromycin, 5) Gentamicin, or 6) Penicillin. Bacteria were then allowed to grow for 36 hours at 37° C. Grow was quantified as positive or negative and individual colonies were selected for future analyses, such as gram staining and 16S rRNA sequencing for speciation. Individual isolates were grown on TSA plates and stored using the Microbank cryogenic system (Pro-lab Diagnostics) at -70° C.
This work has shed insight into the fate of antimicrobial resistance genes in food scraps during composting, as well as establishes a baseline level of on-farm resistance. Additionally, it will streamline future analyses using these techniques as trouble-shooting measures will not need to be repeated. Furthermore, the successful use of a cloud-based bioinformatics platform represents a changing movement towards accessible innovation for smaller labs without dedicated computing or expertise that will be critical in future investigations. Cloud-based bioinformatic methods, while limited in how parameters can be changed, allow laboratories without advanced computing power or resources to perform data-intensive research such as this with a much lower cost and labor input. They have been successfully used in previous research [5, 9]. This reduction in contamination from the previous step (pre-processing) as well as increase in DNA quality here allowed us to prepare a total of 14 samples for sequencing, rather than the 10 samples in the last report. These additional samples allowed for a more complete assessment of the effects of on-farm niche on ARG presence as well as bacterial communities.
A total of 28 samples were sequenced, the 13 experimental samples and 1 trip blank sequenced in duplicate. From these we were able to successfully quantify ARGs in 16 samples and the microbiome of each. Additionally, virulence factors associated with these bacteria were categorized in all 26 experimental samples. The average number of bacteria found (at the species or sub-species level) was 54 distinct hits, followed by an average of 9 virulence genes and 7 antimicrobial resistance genes. Importantly, any carryover from food scraps in both bacterial species and ARGs introduced to the farm appears to be minimal. Upon ordinal analysis, microbiota and resistomes cluster distinctly into external sites and farm niches.
Antimicrobial Resistance Genes
A total of 50 unique resistance genes were found across all samples, most commonly attributed to aminoglycoside and tetracycline resistance. Nine genes for multi-drug efflux genes were found in the samples from the nursing home which did not persist in any of the on-farm samples. Of genes occurring in multiple samples, many were isolated to specific sample types or physical regions on the farm (Figure 1). For example, many genes were only found in the egg wash and shell and shared very few with worm casting samples. This may be due to physical separation, lack of poultry acting as a vector, or a combination of the two.
More specifically, of resistance genes found only three were present in both off-site and on-farm samples. The gene aph6 Id encoding aminoglycoside resistance was found in 9 distinct samples, which has been previously shown to be plasmid mediated and thus easily transferrable between bacterial species [16] Two macrolide resistance genes, mefA and mel, were found in the school, egg wash, and egg shell, but nowhere else on the farm. These genes are known closely related macrolide efflux pumps associated with Streptococcus species [Ambrose which are commonly found on unpasteurized eggs [12].
Significantly, ARGs of clinical concern such as MDR efflux pumps or high-line antibiotic genes were found in site samples (nursing home), but do not appear to integrate onto the farm. This may indicate that while these genes are present in food scraps, they do not confer a competitive advantage to microorganisms throughout the composting process and are therefore mitigated by the current practices on the farm.
Microbial Content
Analysis of the bacterial content and microbiome showed similar trends to that of ARG analysis. Samples of a similar type (i.e. stages of compost or worm casting) or physical location (eggs and food scraps) house a more similar microbiome than more distant samples. Additionally, bacteria commonly thought of as serious human pathogens [18] were found in food scrap collection sites (school, nursing home hospital), but were found much less frequently on the farm and appeared to be mitigated by the composting process as well. As expected, thermophilic bacteria and others previously found to occur in composted samples increased in relative abundance over time in compost samples.
Virulence Factors
Finally, virulence factors found in each sample were also analyzed. Unlike resistance genes, genes related to virulence were identified in every sample. Again, the majority of genes were specific to off-farm or off-farm samples; however, 6 genes were found both on and off farm. Notably, the gene tnpA associated with Proteus mirabilis was found in all four off-farm sites, raw and finished composts, and egg wash. Previous studies indicate that virulence in this species may be plasmid mediated leading to this spread [4].
Phenotypic Resistance
For the functional analysis, we were able to demonstrate that isolates from these samples were functionally resistant to Tetracycline and created an assay system for screening isolates against other antibiotics. This work is an on-going complement to the original proposal and will be published with any academic manuscripts that result from this work. However, even with the single antibiotic tested to date, this is promising data demonstrating that not only is genetic material present in these samples, but that functional copies of these resistance genes exist.
Management Practices
As mentioned in the report from the farm itself [3], current management practices include monitoring for enteric pathogens, inoculation with live compost weekly to ensure biological activity, covering of the feed bin of scraps to reduce unnecessary moisture, and eliminating rodent access to materials. Combined with the natural dynamics of the microbiome, these efforts are likely at least partially responsible for the reduction in both pathogen load and novel resistance genes throughout the composting process.
References:
- Ambrose, K. D., Nisbet, R., & Stephens, D. S. (2005). Macrolide Efflux in Streptococcus pneumoniaeIs Mediated by a Dual Efflux Pump (mel and mef) and Is Erythromycin Inducible. Antimicrobial Agents and Chemotherapy, 49(10), 4203–4209.
- Bengtsson-Palme J, et al. (2014) Shotgun metagenomics reveals a wide array of antibiotic resistance genes and mobile elements in a polluted lake in India. Frontiers in Microbiology, 5:1–14.
- Black Dirt Farm (2017) Feeding Community Food Scraps to Laying Hens in an Active Composting System.
- Burall, LS, Harro JM, Li X, et al. (2004). Proteus mirabilisGenes That Contribute to Pathogenesis of Urinary Tract Infection: Identification of 25 Signature-Tagged Mutants Attenuated at Least 100-Fold . Infection and Immunity, 72(5), 2922–2938.
- Connelly S, Bristol JA, Hubert S, et al. (2017) SYN-004 (ribaxamase), an oral beta-lactamase, mitigates antibioitic-mediated dysbiosis in a procine gut microbiome model. Journal of Applied Microbiology, 123: 66-79
- Dickson RP, et al. (2014) Analysis of culture-dependent versus culture-independent techniques for identification of bacteria in clinically obtained bronchoalveolar lavage fluid. J Clin Microbiol52:3605–3613.
- Doyle, ME (2015) Multidrug-Resistant Pathogens in the Food Supply. Foodborne Pathogens and Disease 12:261–278.
- Jalali S, et al. (2015) Screening Currency Notes for Microbial Pathogens and Antibiotic Resistance Genes Using a Shotgun Metagenomic Approach. PLoS One, 10:e0128711.
- Jumat MR, Hasan NR, Subramanian P, et al. (2017) Membrane Bioreactor-Based Wastewater Treatment Plant in Saudi Arabia: Reduction of Viral Diversity, Load, and Infectious Capacity. Water, 9(534)
- Kim D, Hofstaedter CE, Zhao C, et al. (2017) Optimizing methods and dodging pitfalls in microbiome research. Microbiome, 5(52)
- Minot SS, Krumm N, and Greenfield N (2015) One Codex: A Sensitive and Accurate Data Platform for Genomic Microbial Identification. bioRxiv pre-print
- Moats, W. A. (1980). Classification of bacteria from commercial egg washers and washed and unwashed eggs. Applied and Environmental Microbiology, 40(4), 710–714.
- Musgrove, MT, Jones DR, Nortcutt JK, et al. (2005) Impact of Commercial Processing on the Microbiology of Shell Eggs. Journal of Food Protection 68:11, 2367-2375
- Pehrsson E, et al. (2013) Novel Resistance function uncovered using functional metagenomic investigation of resistance reservoirs. Frontiers in Microbiology, 4 (145):1-11.
- Pitta DW, Dou Z, Kumar S, et al. (2016) Metagenomic Evidence of the Prevalence and Distribution Patterns of Antimicrobial Resistance Genes in Dairy Agroecosystems. Foodborne Pathogens and Disease, 13(6)
- Ramirez MS and Tolmasky ME (2010). Aminoglycoside Modifying Enzymes. Drug Resistance Updates : Reviews and Commentaries in Antimicrobial and Anticancer Chemotherapy, 13(6), 151–171.
- Salter SJ, Cox MJ, Turek EM, et al. (2014) Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology, 12(87)
- Schürch AC and van Schaik W (2017) Challenges and opportunities for whole-genome sequencing-based surveillance of antibiotic resistance. Annals of the New York Academy of Sciences, 1338: 108-120
Tables and Figures.
Figure 1.
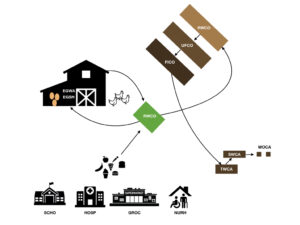
These results provide preliminary evidence that the inclusion and use of food scraps for poultry feed and value-added products such as worm castings is of minimal risk for the spread of novel antibiotic resistance in these settings, the main goal of this work. While future work needs to be conducted, the methods used in this study lay the foundation for more streamlined screening of food scraps and establishes a baseline for expected microbial communities, resistance genes, and virulence factors.
Education & outreach activities and participation summary
Participation summary:
Results from this work were summarized and shared with the participating farmer. This provides initial data to support current farm management practices and a model for other farms looking to incorporate these practices as Act 148 comes into effect. Additionally, this work will generate a manuscript and be presented at the Argonne Soil Metagenomics Conference in November 2017 to experts in the field and researchers that can utilize the methods developed herein for future studies.
Project Outcomes
As shown in the results, while resistance genes are present in the farm environment, specific ARGs from the sites sampled appeared to be mitigated by the composting process. On-farm resistance patterns also clustered by geographic location, and future studies should assess physical containment of substrate as a means to further reducing spread of resistance. Finally, worm castings leaving the farm for consumer use were not found to contain resistance genes using this screening method, so risk of further incorporation into soils is minimal. Furthermore, the microbiome of off vs. on farm samples were distinct, indicating that any potential human pathogens present in raw food scraps are not able to establish a community in the farm environment.
This project has greatly increased our understanding of methodologies, analysis tools, and complementary assays that can be used to assess antimicrobial resistance in soils. It has provided excellent preliminary data to launch future research projects, such as expansion to a greater number of farms and investigation into remediation tools (i.e. phages or inoculants) and will likely result in additional grants and collaborations within our lab group. My individual skills in data analysis and DNA extraction of difficult substrates has been vastly improved through this project, which I will take with me throughout my career. Overall, this project has provided additional evidence for the role of next generation techniques, both sequencing and analysis, in sustainable agriculture and my potential role in it. I hope to use the expertise I gained from this experience to work in a career that bridges animal health, sustainable agriculture, and bioinformatic techniques.
This work has successfully demonstrated that ARGs are present in this environment, as well as that this methodology can be used to study them. However, there are several questions raised to be investigated in future studies. One of the most significant is the mobility of the resistance genes found. While we were able to confirm their presence and functionality, future work should focus on elucidating if these genes are readily transferable between bacterial species. Components of this work may include both genomic and culture-based techniques. For example, more targeted genome sequencing of individual isolates would yield the coverage needed to detect if ARGs were carried within the core or accessory (mobile) genome. The genes occurring in the most samples, resistance gene aph(6)-Id and virulence gene tnpA, are both known to be carried on plasmids suggesting that this may be a common mode of transmission for genes originating in food scraps that are able to establish themselves in the farm environment. In vitro studies could utilize cloning technologies to isolate plasmids and attempt to transfer them between competent strains. This work would provide further insight into the mechanisms of ARG spread, as well as management strategies for prevention.
Secondly, expanding this work to additional farms and time points would continue to grow our understanding of ARG prevalence and transmission dynamics. Expansion to both operations currently composting food scraps and those just starting would highlight any differences between immediate and long-term integration of resistance onto farms and greatly improve management decisions.
Finally, while not covered in the scope of this project, bioinformatic analysis yielded viral sequences present in many samples. These largely include various bacteriophages that could be further investigated as agents of antimicrobial transfer or the mechanism for which naïve bacterial species from food scrap are unable to survive once on farm (i.e. the on farm virome) may have a protective effect and individual phages could be discovered for use in waste facilities as a form of bioremediation or amendment.